Master Regulator in Mitochondria is Critical for Muscle Function & Repair

PHILADELPHIA – When we exercise, our muscles can become fatigued, sore and even damaged with very strenuous activities, like running a marathon. Over time our muscles are able to repair themselves through a complex set of cellular processes within each muscle fiber. New research from the MitoCare Center at Thomas Jefferson University, in collaboration with the Center for Genetic Medicine Research at Children's National Health System in Washington D.C., has identified how MICU1, a protein in the mitochondria, the “powerhouses” of all cells, is critical in the maintenance of muscle size and function, and the repair of damaged muscle fibers. The findings point to a potential role of MICU1 in neuromuscular diseases. The study was published in Cell Reports on October 29th.
The contraction and relaxation of our muscles is dependent on the proper balance of calcium ions inside each muscle fiber. Some of this calcium is taken up by mitochondria, where it is used to boost metabolism and produce energy. The protein MICU1 acts as a master regulator of calcium uptake into the mitochondria.
“Control of calcium transport by MICU1 helps to coordinate muscle fibers and their mitochondria,” says György Hajnóczky, MD, PhD, lead author of the study, professor in the department of pathology, anatomy & cell biology and director of the MitoCare Center. “Disruption of this link prevents the proper communication between mitochondria and the rest of the muscle, making the muscle easier to damage and unable to exert as much force.”
In fact, 50-60 patients without MICU1 have been identified and exhibit neuromuscular symptoms, such as muscle fatigue and weakness. There is an array of neuromuscular and neurodegenerative conditions like muscular dystrophy where calcium dysregulation in mitochondria may be a key player. The underlying mechanisms are unknown.
To tackle this problem, a multicenter collaboration was set up among the groups of Erin Seifert and György Hajnόczky at the MitoCare Center, Jyoti Jaiswal in Washington D.C. and Rita Horvath from the Wellcome Centre for Mitochondrial Research at Newcastle University in Newcastle-Upon-Tyne, UK. First author Valentina Debattisti, a post-doctoral fellow at MitoCare, and her colleagues created a mouse model in which MICU1 is specifically missing in skeletal muscles. They also harvested muscle fibroblasts from patients born without MICU1. This is a novel combined approach that allowed for the study of specific effects of MICU1 deletion in the muscle.
The researchers found that in both the mouse model and patient fibroblasts, loss of MICU1 changes the threshold at which mitochondria takes up calcium, meaning the mitochondria cannot sense the correct level at which to take up calcium. This leads to an imbalance in surrounding levels of calcium in the muscle fiber, preventing its proper function. They found that the muscle fibers produced less contractile force when stimulated, and mice in which MICU1 is deleted show increased fatigue upon exercise and more muscle atrophy, mirroring the symptoms seen in humans without MICU1.
They decided to dig deeper into the possible causes of fatigue and atrophy and honed in on muscle repair. The researchers provoked muscle damage in mice by having them run on a downhill treadmill, which is more strenuous than a running wheel. They then injected a dye that is able to permeate the muscle membrane; the less repair there is, the more dye is present. The researchers found that MICU1-deficient mice had more dye in the muscle, indicating that their ability to undergo muscle membrane repair was compromised. The same was observed in patient fibroblasts, in which they induced damage using a laser. Importantly, when they added MICU1 back to the fibroblasts, the muscle fibers were able to repair themselves.
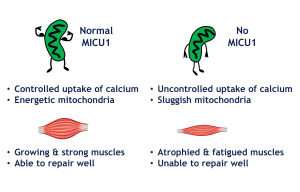
“This means that mitochondrial calcium uptake is important for normal repair of muscle, and this was not known before,” says co-author Dr. Seifert, assistant professor in the department of pathology, anatomy & cell biology and also part of the MitoCare Center. “So boosting this control mechanism could potentially aid in muscle recovery after exercise, especially if the control mechanism is compromised.”
The findings demonstrate a new role for MICU1 in muscle function and establish a direct link between MICU1 and impairments found in neuromuscular diseases, positioning MICU1 as a potential therapeutic target.
“There are so many patients who experience more than normal fatigue when they exercise, which can be debilitating,” says Dr. Hajnóczky. “In many cases it can stem from a mitochondrial disease where calcium transport is disrupted. These patients, as well as those who have other neuromuscular diseases might benefit from a better understanding of MICU1.”
The work was funded by an NIH grants (RO1 GM102724) and from Foundation Leducq. The authors report no conflict of interest.
Article Reference: Valentina Debattisti, Adam Horn, Raghavendra Singh, Erin L. Seifert, Marshall W. Hogarth, Davi A. Mazala, Kai Ting Huang, Rita Horvath, Jyoti K. Jaiswal, and György Hajnóczky, “Dysregulation of Mitochondrial Ca2+ Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1” Cell Reports, DOI: doi.org/10.1016/j.celrep.2019.09.063, 2019
By Karuna Meda
Media Contacts: Edyta Zielinska, 215-955-7359, [email protected]; Karuna Meda, 267-624-4792, [email protected].